Elafibranor v druhé linii léčby primární biliární cholangitidy
Soňa Fraňková Orcid.org 1
+ Pracoviště
Souhrn
Primární biliární cholangitida (PBC) je chronické, imunologicky podmíněné jaterní onemocnění, které ve svém dlouhodobém průběhu vede k destrukci malých žlučovodů, cholestáze, fibróze a cirhóze jater s jaterním selháním. PBC postihuje ve více než 90 % ženy středního věku, většina pacientů je nyní diagnostikována v asymptomatickém stadiu. Diagnóza onemocnění je obvykle stanovena na základě kombinace laboratorních vyšetření, elevace sérové ALP nad 1,5násobek normy trvající déle než 6 měsíců a přítomnosti AMA protilátek v titru 1: 40 nebo vyšším. Typický histologický nález potvrzuje diagnózu, stadium jaterního onemocnění je však nyní možné určit i pomocí neinvazivních metod. Kyselina ursodeoxycholová je v současné době léčbou první volby, v případě intolerance nebo nedostatečné odpovědi na léčbu je možné zahájit léčbu elafibranorem, duálním agonistou PPAR α/δ. Transplantace jater je indikována u pacientů s PBC, kteří dospěli do stadia jaterního selhání i přes podávanou medikamentózní léčbu.
Klíčová slova
primární biliární cholangitida, AMA protilátky, alkalická fosfatáza, kyselina ursodeoxycholová, elafibranor, transplantace jater
Úvod
Primární biliární cholangitida (PBC) je chronické, imunologicky podmíněné jaterní onemocnění, které ve svém dlouhodobém průběhu vede k destrukci malých žlučovodů, cholestáze a progresivní fibróze jater včetně možnosti chronického selhání jater [1]. V minulosti byla obvykle PBC diagnostikována až ve stadiu pokročilého jaterního onemocnění. Choroba byla poprvé popsána v roce 1851 Addisonem a Gullem [2] a následně Hanotem [3] v roce 1876. O sto let později navrhli MacMahon a Thannhauser nové pojmenování „xantomatózní biliární cirhóza“ podle typických xantomů podmíněných akumulací esterů cholesterolu v kůži okolo očí spolu s nálezem zánětlivé destrukce malých žlučovodů vedoucích k biliární cirhóze [4]. Pojmenování primární biliární cirhóza bylo navrženo v roce 1950 Ahrensem a bylo používáno u pacientů s klinicky manifestní chorobou [5]. Již na konci 50. let však bylo zřejmé, že pojmenování není přesné. Britská hepatoložka Sheila Sherlock popsala případy pacientek, které byly v době diagnózy bez klinických příznaků a přežily více než 10 let [6]. Ještě v roce 2009 užívaly doporučené postupy léčby PBC nepřesný název primární biliární cirhóza i přesto, že většina nemocných v době diagnózy cirhózu neměla [7,8]. V květnu roku 2014, u příležitosti konání Monotematické konference o primární biliární cirhóze organizované Evropskou společností pro studium jater (EASL) v Miláně, zažádaly pacientské organizace sdružující pacienty s PBC o změnu pojmenování onemocnění s cílem „odstranit nepřesnost“ a „odejmout stigma cirhózy“, zejména pro časté spojení cirhózy s abúzem alkoholu. Změna pojmenování onemocnění na primární biliární cholangitidu byla následně schválena EASL v listopadu 2014 a Americkou asociací pro studium jater v červnu 2015 [9].
Epidemiologie
Primární biliární cholangitida je onemocnění vzácné, incidence se v jednotlivých studiích významně liší od 0,33 do 5,8/100 000 obyvatel ročně, přesto přispívá významně k mortalitě na jaterní onemocnění [1]. Onemocnění postihuje všechna etnika, významně častěji bělochy a zejména ženy (v poměru 10: 1 k mužům). Průměrný věk v době stanovení diagnózy je 52 let. Velmi vzácně může být onemocnění diagnostikováno již v pubertě či u mladých dospělých. U mužů je diagnóza obvykle stanovena později, mají pokročilejší onemocnění a horší odpověď na léčbu [10,11]. Rizikovým faktorem vzniku PBC je rodinná anamnéza onemocnění, s relativním rizikem 10,5 pro jedince, jejichž sourozenci mají PBC [12], rovněž příbuzní prvního stupně osob s PBC mají vyšší riziko rozvoje onemocnění. Dalšími nezávislými rizikovými faktory jsou přítomnost Sjögrenova syndromu, jiných autoimunních onemocnění (např. onemocnění štítné žlázy, psoriázy), anamnéza cholestázy v graviditě, tonzilektomie, infekce močových cest a vaginální infekce, herpes zoster, cholecystektomie a kouření [13].
Etiopatogeneze
V současné době je PBC považována za orgánově specifické autoimunní onemocnění, které postihuje geneticky disponované jedince [14]. U naprosté většiny nemocných jsou prokazatelné antimitochondriální protilátky (AMA) namířené proti 2-oxodehydrogenáze multienzymových komplexů (2-OADC), s hlavními cíli E2 a E3 podjednotkám pyruvát dehydrogenázového komplexu (PDC-E2 a PDC-E3). Ačkoli je 2-OADC komplex lokalizován na vnitřní mitochondriální membráně všech jaderných buněk, imunologická odpověď u PBC je namířena proti cholangiocytům. PDC-E2 je chybně exprimován na povrchu buněk, zejména malých žlučovodů. Poškození cholangiocytů a duktopenie (destrukcí podmíněný úbytek chlolangiocytů) vede k akumulaci žlučových kyselin, které následně působí toxicky [15]. Dalším mechanizmem je narušení protektivního mechanizmu označovaného jako „bikarbonátový deštník“, který chrání cholangiocyty před toxickým efektem žlučových kyselin udržováním zásaditého pH. Jednou z hlavních složek tohoto systému je na Na+ nezávislá Cl–/HCO3– pumpa (AE2 – anion exchanger 2). Následně dochází k aktivaci eozinofilů a žírných buněk, výsledkem je progresivní granulomatózní zánět vedoucí k duktopenii a zvýšené aktivitě a transformaci hvězdicovitých buněk s produkcí vaziva, onemocnění má rysy syndromu mizejících žlučovodů.
Klinická manifestace
Primární biliární cholangitida postihuje převážně ženy středního věku, pouze asi 10 % postižených jsou muži. Až u 40 % pacientů se onemocnění projeví ve věku > 65 let, klinický obraz se však neliší od pacientů diagnostikovaných v mladších věkových kategoriích [10].
Klinický obraz při stanovení diagnózy se zásadně změnil od prvního popisu onemocnění v roce 1851. Většina pacientů je nyní diagnostikována v asymptomatickém stadiu, zejména v západních zemích (až 85 %), na základě náhodného nálezu elevace aktivity jaterních enzymů, zejména ALP (alkalické fosfatázy). Dalšími typickými příznaky jsou únava a svědivka, která se může manifestovat roky před stanovením diagnózy. Ikterus je příznakem pozdním a je spojen s nepříznivou prognózou. Bolest v pravém podžebří udává asi 10 % nemocných [16].
Únava je přítomna až u 85 % nemocných s PBC a přibližně polovina nemocných ji vnímá jako nejzávažnější příznak, který velmi nepříznivě ovlivňuje kvalitu života [17]. Nemocní trpí poruchami spánku a nadměrnou spavostí ve dne. Pacienti mají nižší variabilitu tepové srdeční frekvence a jsou hypotenzní. Dalšími faktory, které mohou k únavě přispět, jsou deprese, spánková deprivace, nežádoucí účinky podávané medikace, anemie a hypofunkce štítné žlázy.
Pruritus je definován jako nepříjemný pocit nutící pacienta ke škrábání kůže. Je častým steskem nemocných s PBC, vyskytuje se až u 70 % pacientů [18]. Příčina pruritu byla po dlouhou dobu neznámá, v současnosti se má za to, že je za něj zodpovědná osa lipopolysacha- rid–autotaxin. Autotaxin je enzym zodpovědný za vznik lipopolysacharidu z jeho prekurzoru lipofosfatidylcholinu. Sérová koncentrace autotaxinu koreluje s intenzitou pruritu a s odpovědí na jeho léčbu. Ačkoli některé studie stanovily aktivitu alkalické fosfatázy a hodnotu Mayo risk skóre jako nezávislé prediktory pruritu, dle jiných neexistuje jasná korelace mezi biochemickými markery cholestázy, stadiem onemocnění a pruritem [19,20]. Pruritus obvykle postihuje kůži celého těla, je intermitentní a významně snižuje kvalitu života. Vede k depresi a poruchám spánku. Neztišitelný pruritus sám o sobě může být indikací k transplantaci jater bez ohledu na pokročilost jaterní dysfunkce.
Pouze u malé části pacientů s PBC jsou přítomny známky portální hypertenze v době stanovení diagnózy. Klinické známky portální hypertenze jsou obdobné jako u ostatních pacientů s jaterní cirhózou.
Extrahepatální komplikace
Osteoporóza komplikuje PBC u 20–40 % pacientů a představuje dvojnásobné riziko fraktur, zejména v oblasti páteře, kyčlí a předloktí. Rizikovými faktory osteoporózy jsou vyšší věk, nízký BMI (≤ 24 kg/m2) a pokročilé stadium onemocnění. Při progresi onemocnění a cholestáze dochází k malabsorbci v tucích rozpustných vitaminů A, D, E a K. Riziko malabsorbce vitaminů stoupá s pokročilostí choroby a snižující se sérovou koncentrací cholesterolu a albuminu [21].
Hyperlipidemie je přítomna až u 85 % pacientů s PBC při diagnóze onemocnění. Je přítomna významná elevace lipoproteinů s vysokou denzitou (HDL), zatímco elevace lipoproteinů s nízkou (LDL) a velmi nízkou denzitou (VLDL) není tak výrazná [22]. S progresí choroby klesají hodnoty HDL, zatímco hodnoty LDL zůstávají zvýšené z důvodu snížení množství LDL receptorů na poškozených hepatocytech. Xantelesmata a xantomy jsou častým nálezem u pacientů s PBC. Dosud nebyla prokázána jasná korelace mezi hyperlipidemií provázející PBC a kardiovaskulárním rizikem. Léčba hyperlipidemie má být proto zahájena s ohledem na ostatní rizikové faktory kardiovaskulárních onemocnění u jednotlivých pacientů [23].
Primární biliární cholangitida je spojena s mnoha dalšími autoimunními onemocněními, jako je Sjögrenův syndrom (až 70 %), thyroiditida (15 %), sklerodermie a CREST syndrom, revmatoidní artritida, systémový lupus erythematodes a celiakie. Pacienti s PBC mají rovněž vyšší riziko vzniku hepatocelulárního karcinomu, zejména v závislosti na pokročilosti onemocnění a na chybění odpovědi na terapii kyselinou ursodeoxycholovou [24].
Diagnóza
Diagnóza onemocnění je obvykle stanovena na základě kombinace laboratorních vyšetření, elevace sérové ALP > 1,5násobek normy trvající déle než 6 měsíců a přítomnosti AMA protilátek v titru 1: 40 nebo vyšším [14,25]. Typický histologický nález floridní nonsupurativní duktální léze s tvorbou nekaseifikujících granulomů potvrzuje diagnózu PBC, jaterní biopsie ale není pro stanovení diagnózy nezbytná při splnění dvou výše uvedených kritérií. AMA negativní pacienti se suspekcí na PBC by k jaterní biopsii být indikováni měli. V diferenciální diagnóze pak zvažujeme zejména extrahepatální obstrukci žlučových cest, choledocholitiázu, tumory žlučovodů, primární sklerozující cholangitidu, polékové jaterní poškození, autoimunní hepatitidu, sarkoidózu či IgG4 asociovanou cholangitidu [14].
Ačkoli při PBC mohou být zvýšené i hodnoty jaterních aminotransferáz, laboratornímu obrazu dominuje cholestáza, elevace ALP a GGT. Standardně je rovněž přítomna elevace imunoglobulinů ve třídě IgM a hyperlipidemie. Elevace koncentrace bilirubinu, snížená hodnota albuminu a prodloužení protrombinového času jsou známkami pokročilé jaterní dysfunkce. Přítomnost AMA protilátek je základním znakem diagnózy PBC. AMA protilátky mohou být negativní až u 10 % pacientů při průkazu nepřímou imunofluorescencí, vyšší senzitivity dosáhneme pomocí vyšetření metodou ELISA. K definitivnímu potvrzení diagnózy AMA-negativní PBC je nezbytné provedení jaterní biopsie a její zhodnocení zkušeným patologem. V obecné populaci nacházíme pozitivitu AMA asi u 0,5 % asymptomatických jedinců, PBC z nich vyvine méně než 10 %. Antinukleární (ANA) protilátky jsou přítomny až v 50 % případů u nemocných s PBC, pro PBC jsou typické anti-sp100, anti-gp210.
Vyšetření ultrazvukem je základní zobrazovací metodou a vyloučí obstrukci žlučových cest, přinese navíc informace o struktuře jater, přítomnosti portální hypertenze, velikosti sleziny, vyloučí ascites či ložiskové postižení jater.
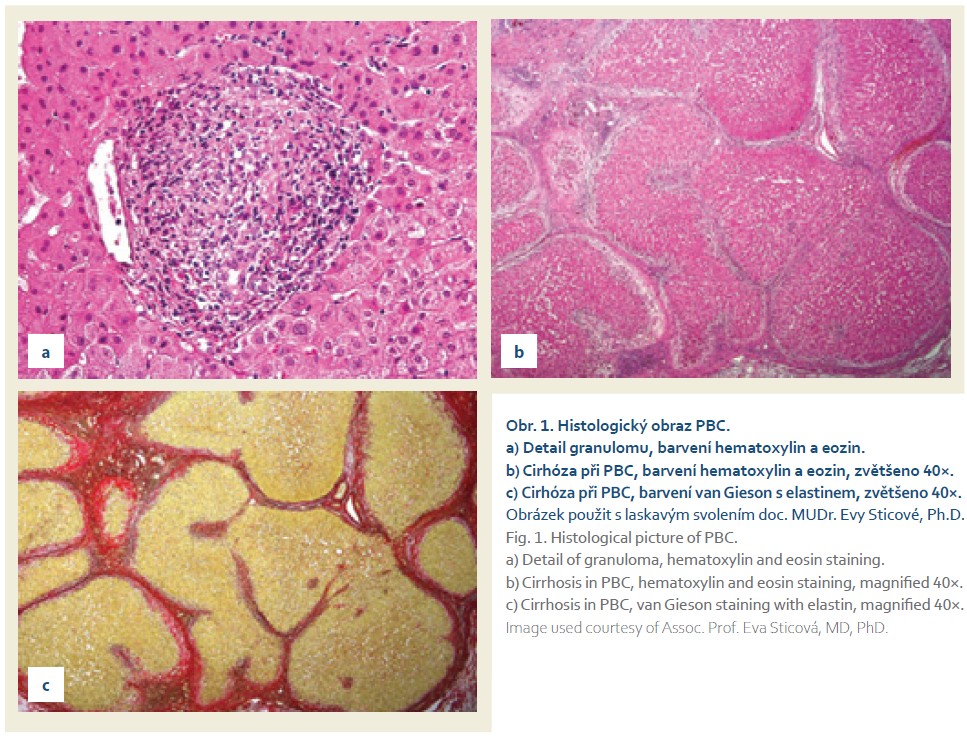
Se zlepšujícími se možnostmi laboratorní diagnostiky onemocnění a při možnosti hodnocení progrese onemocnění pomocí neinvazivních metod hodnocení jaterní fibrózy klesl význam jaterní biopsie v diagnostice PBC. Při PBC jsou postiženy žlučovody průměru < 100 μm. V histologické klasifikaci užíváme dva základní stagingové systémy: klasifikaci Ludwigovu a Scheuerovu, oba klasifikační systémy jsou si podobné [26]. Stadium I popisuje zánět a nehnisavou destrukci žlučovodů. Infiltráty obsahují lymfocyty, plazmatické buňky, eozinofily. Proces je izolován na portální pole. Jsou přítomny epiteloidní nekaseifikující granulomy v blízkosti poškozených žlučovodů (obr. 1a). Ve stadiu II je již přítomna interface aktivita. Obě tato stadia jsou časná. Stadium III odpovídá septální fibróze, stadium IV pak cirhóze jater (obr. 1b, c). Histologické skóre podle Nakanumy zahrnuje navíc stupeň aktivity cholangitidy (0–3) a aktivitu hepatitickou (0–3). Toto skóre koreluje s prognózou pacienta [27].
Progrese onemocnění a prognóza
Onemocnění má dlouhodobý průběh, můžeme jej rozdělit do čtyř stadií: preklinického, asymptomatického, symptomatického a stadia jaterního selhání. PBC je vysoce heterogenní onemocnění a riziko a rychlost progrese onemocnění se u jednotlivých pacientů významně liší. Prognóza u pacientů s již vyvinutou cirhózou může být stanovena na základě Child-Pughova [28] nebo MELD (model for end-stage liver disease) skóre [29]. V současnosti je většina pacientů s PBC diagnostikována ve velmi časných fázích choroby s výhledem zpomalení progrese onemocnění léčbou, a nikdy tak nedospějí do stadia chronického selhání jater. Z tohoto důvodu je důležitá schopnost zhodnotit riziko progrese onemocnění u jednotlivých pacientů. Stupeň fibrózy jater zhodnocený jaterní biopsií nebo neinvazivními metodami (tranzientní nebo MR elastografie) [30] adekvátně koreluje s pokročilými stupni PBC a komplikacemi vyplývajícími z portální hypertenze, ze stejné indikace lze použít invazivní měření portosystémového gradientu [31].
Progrese PBC mezi jednotlivými stadii onemocnění je vysoce variabilní, a je tedy nutné využívat dynamických modelů ke stanovení střednědobé a dlouhodobé prognózy u jedinců diagnostikovaných ve velmi časných fázích choroby. K určení prognózy byly užívány četné biochemické i klinické parametry, zejména věk manifestace < 50 let byl považován za indikátor špatné prognózy. Stejně tak zvýšená koncentrace bilirubinu, žloutenka, únava a pruritus v době stanovení diagnózy byly spojovány s horší prognózou [32,33]. Pravděpodobně nejlepším prognostickým markerem je sérová aktivita ALP, jejíž hodnota v multivariantní analýze 4 845 pacientů s PBC úzce korelovala s rizikem úmrtí a nutností transplantace jater [34]. Výhoda ALP tkví ve schopnosti predikce právě v časných fázích onemocnění, nezávisle na příznacích, věku, pohlaví, stadiu choroby a podávané léčbě. Prognostická hodnota nabývá na významu rovněž v souvislosti se změnou koncentrace ALP při léčbě kyselinou ursodeoxycholovou, která v dlouhodobém horizontu dokáže ovlivnit průběh onemocnění.
Léčba
Kyselina ursodeoxycholová
Kyselina ursodeoxycholová (UDCA) byla prvním lékem registrovaným k léčbě PBC [35]. Dávka 13–15 mg/kg má vyšší účinnost než podávání dávek nižších (5–7 mg/kg). V současné době je v léčbě PBC terapií první linie [14,36]. Nově diagnostikovaní pacienti by měli zahájit léčbu i v časných fázích onemocnění, se zhodnocením efektu rok po zahájení léčby, aby bylo možno posoudit jejich dlouhodobou prognózu. Při léčbě dochází k poklesu hodnot sérového bilirubinu, ALP, GGT, cholesterolu i imunoglobulinů třídy IgM. Spolu s laboratorními nálezy se zlepšuje i histologický nález: léčba signifikantně zpomaluje progresi fibrózy, snižuje portální zánět a zlepšuje duktulární proliferaci. Riziko komplikací vyplývající z onemocnění je nižší u pacientů léčených UDCA v dávce > 13 mg/kg a den než u nemocných léčených dávkou nižší. Přesto i nemocní užívající denní dávku < 13 mg/kg mají šanci na přežití bez transplantace signifikantně vyšší než pacienti, kteří nejsou léčeni vůbec [37].
Účinek UDCA na symptomy, jako jsou únava, pruritus a kostní choroba, nemusí být významný. Léčba UDCA jednoznačně zlepšuje přežití pacientů s PBC a snižuje nutnost indikace k transplantaci jater. Mechanizmus účinku UDCA u PBC je komplexní. Jedná se o hydrofilní žlučovou kyselinu bez cytotoxického efektu na buněčné membrány, podání UDCA tak chrání cholangiocyty a má rovněž účinek choleretický. Stimulace sekrece žluči je dosaženo pomocí up-regulace syntézy, apikální inzerce a aktivace pumpy žlučových kyselin (BSEP). Další účinek UDCA je imunomodulační, dokáže ovlivnit aberantní expresi molekul HLA I na hepatocytech a modulovat sekreci cytokinů periferními monocyty.
Pacienti, u nichž léčba UDCA vede k poklesu nebo normalizaci ALP, mají signifikantně lepší prognózu ve srovnání s pacienty bez odpovědi na léčbu. Tento fakt vedl k zavedení prognostických modelů dlouhodobé prognózy, které predikují přežití a nutnost transplantace při léčbě UDCA. Většina těchto modelů hodnotí odpověď na UDCA rok po zahájení léčby. Ve studii Parese et al. bylo prokázáno, že pacienti, kteří po roce léčby dosáhnou normalizace nebo alespoň snížení iniciální hodnoty ALP o 40 %, mají srovnatelné přežití s kontrolní zdravou populací a jednoznačně lepší přežití oproti přežití predikovanému dle tzv. Mayo risk skóre [38]. Tento model je obecně znám jako „Barcelonská kritéria“ [38]. V práci publikované v roce 2008 Corpechotem et al. [39] byly definovány další parametry, jejichž dosažení vede k signifikantně lepšímu přežití při léčbě UDCA. Tato kritéria jsou známá jako „Pařížská kritéria“. Hodnoty sérového bilirubinu ≤ 17 μmol/l, ALP ≤ trojnásobku normálních hodnot a AST ≤ dvojnásobku normálních hodnot po roce léčby identifikují pacienty s dobrou prognózou s nízkým rizikem nutnosti transplantace jater či úmrtí na jaterní onemocnění. UK-PBC risk skóre [11] identifikovalo v multivariantní analýze jako zásadní faktory prognózy albumin a počet trombocytů při stanovení diagnózy a změnu ALP, bilirubinu a ALT po 12 měsících léčby UDCA. UK-PBC risk skóre v současné době představuje nejpřesnější prediktivní model. Predikce prognózy na základě Global PBC study group na téměř 5 tisících pacientech je založena na normalizaci bilirubinu a poklesu ALP pod dvojnásobek normy po roce léčby UDCA [36,40].
„Torontská kritéria“ hodnotí přítomnost duktopenie v jaterní biopsii a pokles ALP při léčbě UDCA. Pacienti, u nichž klesla po 2 letech léčby aktivita ALP < 1,67násobek normy, měli nižší riziko progrese fibrózy jater [41]. Pokles ALP < 1,67násobek normy je v současné době nejužívanějším kritériem hodnocení odpovědi na léčbu UDCA, nicméně v naprosté většině studií se kritérium odpovědi hodnotí již po roce terapie [42].
Pacienti, u nichž dojde nejen k poklesu, ale úplné normalizaci ALP při léčbě UDCA (tzv. deep response, hluboká odpověď), mají přežití bez komplikací vyplývajících z jaterního onemocnění jednoznačně nejvyšší. Tento fakt je dobře dokumentován ve studii Murillo Perez et al. z roku 2020: pacienti, kteří docílili normalizace ALP, měli signifikantně lepší 10leté přežití ve srovnání s nemocnými, kteří dosáhli aktivity ALP mezi hodní hranicí a 1,67násobku normy (93,2 %; resp. 86,1 %) [36].
Přínos kompletní normalizace ALP potvrzuje i studie provedená v 24 centrech na 1 047 pacientech: pacienti, kteří dosáhli úplné normalizace ALP, měli signifikantně nižší riziko komplikací spojených s onemocněním jater ve srovnání s pacienty, kteří sice na léčbu odpověděli ve smyslu poklesu ALP < 1,67násobek normy, ale normalizace nedosáhli, včetně pacientů s tuhostí jater ≥ 10,0 kPa hodnocenou tranzientní elastografií [43].
Přibližně 40 % pacientů však na léčbu UDCA neodpoví adekvátně či léčbu netoleruje, a proto musí být ukončena [44]. Tito pacienti jsou ohroženi progresí onemocnění [37], a má jim proto být nabídnuta léčba druhé linie.
Elafibranor
Elafibranor a jeho hlavní aktivní metabolit (GFT1007) jsou duální agonisté receptorů aktivovaných proliferátory peroxisomů (PPAR) α a δ. PPAR α a δ jsou klíčovými regulátory homeostázy žlučových kyselin, zánětu a fibrózy [45]. Jejich aktivace snižuje produkci žlučových kyselin, zvyšuje jejich detoxifikaci a ovlivňuje jejich výdej, což vede ke snížení jejich toxicity, a tedy snížení rizika poškození cholangiocytů a hepatocytů. Aktivace PPR α a δ má rovněž účinek protizánětlivý, působí na několika místech zánětlivé kaskády ovlivněním dráhy NF-k B a BCL6 [46].
Klinická účinnost elafibranoru byla hodnocena ve studii GFT505B-319-1 (ELATIVE), randomizované, dvojitě zaslepené, placebem kontrolované studii fáze 3 [47]. Do studie bylo zařazeno 161 dospělých pacientů s PBC, kteří měli nedostatečnou odpověď na UDCA nebo ji netolerovali. Podávaná dávka elafibranoru byla 80 mg jednou denně, doba léčby byla nejméně 52 týdnů. Do studie nebyli zařazeni pacienti s dekompenzovanou jaterní cirhózou.
Průměrný věk účastníků studie byl 57,1 roku, průměrná hmotnost 70,8 kg. Studijní populaci tvořily převážně ženy (96 %). Průměrná aktivita ALP na počátku léčby byla 321,9 IU/l (5,37 μkat/l), 35 % účastníků studie mělo pokročilé onemocnění jater definované jako tuhost jater při měření tranzientní elastografií ≥ 10 kPa nebo přemosťující fibróza nebo cirhóza v jaterní biopsii. Většina účastníků (95 %) dostávala elafibranor v kombinaci s UDCA, pouze 5 % nemocných bylo léčeno monoterapií elafibranor/placebo. Celkem 41 % nemocných mělo na začátku terapie středně těžkou nebo těžkou svědivku.
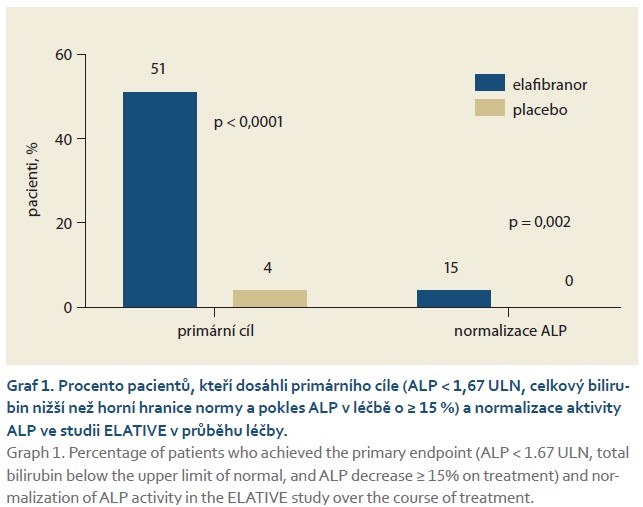
Primárním cílem byl pokles cholestázy v 52. týdnu studie definovaný jako složený cílový parametr: ALP < 1,67 ULN, celkový bilirubin nižší než horní hranice normy a pokles ALP v léčbě o ≥ 15 %. Klíčovými sekundárními cíli byla normalizace aktivity ALP v 52. týdnu terapie a změna intenzity pruritu do 24. a 52. týdne na základě WI-NRS škály (the worst itch numeric rating scale) u pacientů se středně těžkou a těžkou svědivkou na začátku terapie.
Významný pokles ALP oproti výchozím hodnotám byl zaznamenán již ve 4. týdnu a trval až do týdne 52 (graf 1). Primárního cíle v týdnu 52 dosáhlo celkem 51 % léčených pacientů ve skupině s elafibranorem oproti 4 % pacientů léčených placebem (p < 0,0001). Sekundárního cíle, normalizace ALP, dosáhlo v 52. týdnu 15 % nemocných léčených elafibranorem (p = 0,002 ve srovnání s placebem) (graf 2).

Mezi pacienty se středně těžkým a těžkým pruritem byl pozorován pokles skóre WI-NRS ve 24. i 52. týdnu léčby, nicméně hodnoty nedosáhly statistické významnosti. Elafibranor rovněž prokázal příznivý vliv na parametry lipidového spektra. Snížení koncentrace cholesterolu, lipoproteinů s velmi nízkou denzitou a triglyceridů bylo vyšší než u pacientů léčených placebem.
Nejčastěji hlášenými nežádoucími účinky souvisejícími s léčbou elafibranorem ve studii ELATIVE [46,47] vyskytujícími se u > 10 % účastníků a s vyšší incidencí než ve skupině s placebem byly bolest břicha, průjem, nauzea a zvracení. Nejčastějším nežádoucím účinkem, který vedl k ukončení léčby, byla zvýšená aktivita kreatinkinázy (CK) v krvi (3,7 %). Toto zvýšení nebylo zaznamenáno u nikoho ve větvi s placebem. U dvou ze čtyř pacientů s elevací CK byla hodnota vyšší než 5násobek horní hranice normy. Všechny případy byly nezávažné, mírné nebo střední intenzity, dva pacienti měli současně myalgii.
Elafibranor se podává v dávce 80 mg jednou denně, v kombinaci s UDCA u dospělých pacientů s nedostatečnou odpovědí na UDCA. U nemocných, kteří terapii UDCA netolerují, se podává elafibranor v monoterapii. U starších pacientů není nutná úprava dávky, ta rovněž není nutná u pacientů s poruchou funkce ledvin. Elafibranor lze rovněž podávat bez úpravy dávky u pacientů s jaterní cirhózou a lehkou či středně těžkou jaterní dysfunkcí (třída A a B Child-Pughovy klasifikace), u pacientů s těžkou jaterní dysfunkcí se podání elafibranoru nedoporučuje [46]. Na základě studií in vitro i in vivo nejsou očekávány klinicky relevantní lékové interakce s jakýmikoli jinými léčivými přípravky.
Elafibranor byl schválen v září 2024 Evropskou agenturou pro léčivé přípravky (EMA) k léčbě PBC v kombinaci s UDCA u pacientů s nedostatečnou odpovědí na UDCA nebo v monoterapii u pacientů UDCA netolerujících.
Léčba extrahepatálních komplikací a pruritu
Pruritus patří mezi nejčastější příznaky provázející PBC. Značně ovlivňuje kvalitu života a v případech rezistentních ke standardní terapii může být i indikací k transplantaci jater. Cholestyramin je léčbou pruritu první linie v úvodní celkové dávce 4 g denně. Užití cholestyraminu musí být odděleno od ostatní medikace, zejména UDCA, o 2–4 hod. V případě neúčinnosti této léčby by další volbou mohlo být podání rifampicinu [14,48]. Jako třetí linii lze použít léky ze skupiny perorálních opiodních antagonistů (např. naltrexon) [25].
Nemocné s PBC je třeba pravidelně sledovat s ohledem na decifit vitaminů rozpustných v tucích a tyto vitaminy mají být adekvátně suplementovány. Dosud nebyla prokázána souvislost mezi hyperlipidemií při PBC a zvýšeným kardiovaskulárním rizikem. Léčba hyperlipidemie má být zahájena po posouzení individuálních rizikových faktorů a rodinné anamnézy.
Velmi častou komplikací PBC je osteoporóza. Vysoké riziko je zvláště u nemocných s pokročilou jaterní chorobou a výraznou cholestázou. Při stanovení diagnózy PBC má být u všech pacientů provedena kostní denzitometrie, kontrolní vyšetření pak mají následovat v 2–3letých intervalech [25]. Včasné zahájení kostní choroby je zásadní, suplementujeme vápník v dávce 1 500 mg denně a vitamin D (1 000 IU denně). U závažnějších stavů je indikována léčba bisfosfonáty (zejména alendronát), které jsou i u nemocných s PBC bezpečné, zvýšené opatrnosti při perorální léčbě bisfosfonáty je třeba u nemocných s jícnovými varixy [14].
Transplantace jater
Transplantace jater představuje konečnou léčbu PBC u pacientů, kteří dospějí do stadia jaterního selhání i přes podávanou medikamentózní léčbu. Pravidla indikace k transplantaci jsou shodná jako u pacientů s jaterním selháním při cirhóze jiné etiologie či s malým hepatocelulárním karcinomem [49]. Neztišitelný pruritus a extrémní únava mohou být vzácně indikací k transplantaci jater i při kompenzované jaterní cirhóze. Počet transplantací jater pro PBC celosvětově klesá díky léčbě UDCA, přežití nemocných indikovaných k transplantaci jater je excelentní, 5leté přežití se pohybuje mezi 78 a 90 % [50–52].
Závěr
Primární biliární cholangoitida je chronické cholestatické autoimunitní jaterní onemocnění, u neléčených nemocných s vysokým rizikem progrese do jaterní cirhózy. Diagnóza je založena na přítomnosti cholestatického laboratorního obrazu, průkazu AMA protilátek nebo typickém histologickém nálezu. Lékem první volby je UDCA spolu s léčbou symptomů (zejména pruritu) a prevence a léčba kostní choroby. V případě adekvátní odpovědi na léčbu UDCA je prognóza nemocných dobrá. U non-respondérů či u pacientů, kteří léčbu UDCA netolerují, můžeme podat v druhé linii léčby elafibranor. U nemocných s dekompenzovanou jaterní cirhózou je nutno zvážit indikaci k transplantaci jater.
ORCID autorky
S. Fraňková 0000-0002-1462-5920.
Doručeno/Submitted: 27. 3. 2025
Přijato/Accepted: 2. 4. 2025
Korespondenční autorka
doc. MUDr. Soňa Fraňková, Ph.D.
Klinika hepatogastroenterologie
Institut klinické a experimentální medicíny
Vídeňská 1958/9
140 21 Praha 4
sona.frankova@ikem.cz
Pro přístup k článku se, prosím, registrujte.
Výhody pro předplatitele
Výhody pro přihlášené
Literatura
1. Eksteen B. Predicting risk in primary biliary cholangitis. Hepatology 2016; 63(3): 697–699. doi: 10.1002/hep.28377.
2. Addison T, Gull W. On a certain affection of the skin-vitiligoidea-α plana, ß tuberosa. Guy’s Hosp Rep 1851; 7: 265–276.
3. Hanot V. Etude sur une forme de cirrhose hypertrophique du foie (cirrhose hypertrophique avec ictere chronique). Paris: J.-B. Baillere 1876.
4. Mac MH, Thannhauser SJ. Xanthomatous biliary cirrhosis; a clinical syndrome. Ann Intern Med 1949; 30(1): 121–179. doi: 10.7326/0003-4819-30-1-121.
5. Ahrens EH, Payne MA, Kunkel HG et al. Primary biliary cirrhosis. Medicine 1950; 29 (4): 299–364. doi: 10.1097/00005792-195012000-00002.
6. Sherlock S. Primary billiary cirrhosis (chronic intrahepatic obstructive jaundice). Gastroenterology 1959; 37: 574–586.
7. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: management of cholestatic liver diseases. J Hepatol 2009; 51(2): 237–267. doi: 10.1016/ j.jhep.2009.04.009.
8. Lindor KD, Gershwin ME, Poupon R et al. Primary biliary cirrhosis. Hepatology 2009; 50(1): 291–308. doi: 10.1002/hep.22906.
9. Beuers U, Gershwin ME, Gish RG et al. Changing nomenclature for PBC: from “cirrhosis” to “cholangitis”. J Hepatol 2015; 63(5): 1285–1287. doi: 10.1016/j.jhep.2015.06.031.
10. Kim WR, Lindor KD, Locke GR et al. Epidemiology and natural history of primary biliary cirrhosis in a US community. Gastroenterology 2000; 119(6): 1631–1636. doi: 10.1053/gast.2000.20197.
11. Carbone M, Sharp SJ, Flack S et al. The UK-PBC risk scores: derivation and validation of a scoring system for long-term prediction of end-stage liver disease in primary biliary cholangitis. Hepatology 2016; 63(3): 930–950. doi: 10.1002/hep.28017.
12. Jones DE, Watt FE, Metcalf JV et al. Familial primary biliary cirrhosis reassessed: a geographically-based population study. J Hepatol 1999; 30(3): 402–407. doi: 10.1016/s0168-8278(99)80097-x.
13. Gershwin ME, Selmi C, Worman HJ et al. Risk factors and comorbidities in primary biliary cirrhosis: a controlled interview-based study of 1032 patients. Hepatology 2005; 42(5): 1194–1202. doi: 10.1002/hep.20907.
14. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: the diag- nosis and management of patients with primary biliary cholangitis. J Hepatol 2017; 67(1): 145–172. doi: 10.1016/j.jhep.2017.03.022.
15. Salunga TL, Cui ZG, Shimoda S et al. Oxidative stress-induced apoptosis of bile duct cells in primary biliary cirrhosis. J Autoimmun 2007; 29(2–3): 78–86. doi: 10.1016/j.jaut.2007.04.002.
16. Kumagi T, Onji M. Presentation and diagnosis of primary biliary cirrhosis in the 21st century. Clin Liver Dis 2008; 12(2): 243–259. doi: 10.1016/j.cld.2008.02.014.
17. Jones DE, Bhala N, Burt J et al. Four year follow-up of fatigue in a geographically defined primary biliary cirrhosis patient cohort. Gut 2006; 55(4): 536–541. doi: 10.1136/gut.2005.080317.
18. Prince M, Chetwynd A, Newman W et al. Survival and symptom progression in a geographically based cohort of patients with primary biliary cirrhosis: follow-up for up to 28 years. Gastroenterology 2002; 123(4): 1044–1051. doi: 10.1053/gast.2002.36027.
19. Talwalkar JA, Souto E, Jorgensen RA et al. Natural history of pruritus in primary biliary cirrhosis. Clin Gastroenterol Hepatol 2003; 1(4): 297–302.
20. Poupon R, Chazouilleres O, Balkau B et al. Clinical and biochemical expression of the histopathological lesions of primary biliary cirrhosis. UDCA-PBC Group. J Hepatol 1999; 30(3): 408–412. doi: 10.1016/s0168-8278(99)80098-1.
21. Phillips JR, Angulo P, Petterson T et al. Fat-soluble vitamin levels in patients with primary biliary cirrhosis. Am J Gastroenterol 2001; 96(9): 2745–2750. doi: 10.1111/j.1572-0241. 2001.04134.x.
22. Crippin JS, Lindor KD, Jorgensen R et al. Hypercholesterolemia and atherosclerosis in primary biliary cirrhosis: what is the risk? Hepatology 1992; 15(5): 858–862. doi: 10.1002/hep.1840150518.
23. Sorokin A, Brown JL, Thompson PD. Primary biliary cirrhosis, hyperlipidemia, and atherosclerotic risk: a systematic review. Atherosclerosis 2007; 194(2): 293–299. doi: 10.1016/j.atherosclerosis.2006.11.036.
24. Trivedi PJ, Lammers WJ, van Buuren HR et al. Stratification of hepatocellular carcinoma risk in primary biliary cirrhosis: a multicentre international study. Gut 2016; 65(2): 321–329. doi: 10.1136/gutjnl-2014-308351.
25. Lindor KD, Bowlus CL, Boyer J et al. Primary biliary cholangitis: 2018 practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2019; 69(1): 394–419. doi: 10.1002/hep.30145.
26. Burt AD. Primary biliary cirrhosis and other ductopenic diseases. Clin Liver Dis 2002; 6(2): 363–380. doi: 10.1016/s1089-3261(02)00010-7.
27. Nakanuma Y, Zen Y, Harada K et al. Application of a new histological staging and grading system for primary biliary cirrhosis to liver biopsy specimens: interobserver agreement. Pathol Int 2010; 60(3): 167–174. doi: 10.1111/j.1440-1827.2009.02500.x.
28. Pugh RN, Murray-Lyon IM, Dawson JL et al. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg 1973; 60(8): 646–649. doi: 10.1002/bjs.1800600817.
29. Kamath PS, Wiesner RH, Malinchoc M et al. A model to predict survival in patients with end-stage liver disease. Hepatology 2001; 33 (2): 464–470. doi: 10.1053/jhep.2001.22172.
30. Corpechot C, Carrat F, Poujol-Robert A et al. Noninvasive elastography-based assessment of liver fibrosis progression and prognosis in primary biliary cirrhosis. Hepatology 2012; 56 (1): 198–208. doi: 10.1002/hep.25599.
31. Huet PM, Vincent C, Deslaurier J et al. Portal hypertension and primary biliary cirrhosis: effect of long-term ursodeoxycholic acid treatment. Gastroenterology 2008; 135 (5): 1552–1560. doi: 10.1053/j.gastro.2008.07.019.
32. Dickson ER, Grambsch PM, Fleming TR et al. Prognosis in primary biliary cirrhosis: model for decision making. Hepatology 1989; 10(1): 1–7. doi: 10.1002/hep.1840100102.
33. Grambsch PM, Dickson ER, Kaplan M et al. Extramural cross-validation of the Mayo primary biliary cirrhosis survival model establishes its generalizability. Hepatology 1989; 10(5): 846–850. doi: 10.1002/hep.1840100516.
34. Lammers WJ, van Buuren HR, Hirschfield GM et al. Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: an international follow-up study. Gastroenterology 2014; 147(6): 1338.e5–1349.e5. doi: 10.1053/ j.gastro.2014.08.029.
35. Poupon RE, Balkau B, Eschwege E et al. A multicenter, controlled trial of ursodiol for the treatment of primary biliary cirrhosis. UDCA-PBC study group. N Engl J Med 1991; 324(22): 1548–1554. doi: 10.1056/NEJM199105 303242204.
36. Murillo Perez CF, Harms MH, Lindor KD et al. Goals of treatment for improved survival in primary biliary cholangitis: treatment target should be bilirubin within the normal range and normalization of alkaline phosphatase. Am J Gastroenterol 2020; 115(7): 1066–1074. doi: 10.14309/ajg.0000000000000557.
37. Harms MH, van Buuren HR, Corpechot C et al. Ursodeoxycholic acid therapy and liver transplant-free survival in patients with primary biliary cholangitis. J Hepatol 2019; 71(2): 357–365. doi: 10.1016/j.jhep.2019.04.001.
38. Pares A, Caballeria L, Rodes J. Excellent long-term survival in patients with primary biliary cirrhosis and biochemical response to ursodeoxycholic acid. Gastroenterology 2006; 130(3): 715–720. doi: 10.1053/j.gastro.2005.12.029.
39. Corpechot C, Abenavoli L, Rabahi N et al. Biochemical response to ursodeoxycholic acid and long-term prognosis in primary biliary cirrhosis. Hepatology 2008; 48(3): 871–877. doi: 10.1002/hep.22428.
40. Lammers WJ, Hirschfield GM, Corpechot C et al. Development and validation of a scoring system to predict outcomes of patients with primary biliary cirrhosis receiving ursodeoxycholic acid therapy. Gastroenterology 2015; 149(7): 1804.e4–1812.e4. doi: 10.1053/ j.gastro.2015.07.061.
41. Kumagi T, Guindi M, Fischer SE et al. Baseline ductopenia and treatment response predict long-term histological progression in primary biliary cirrhosis. Am J Gastroenterol 2010; 105(10): 2186–2194. doi: 10.1038/ajg.2010.216.
42. Nevens F, Andreone P, Mazzella G et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med 2016; 375(7): 631–643. doi: 10.1056/NEJMoa1509840.
43. Corpechot C, Lemoinne S, Soret PA et al. Adequate versus deep response to ursodeoxycholic acid in primary biliary cholangitis: to what extent and under what conditions is normal alkaline phosphatase level associated with complication-free survival gain? Hepatology 2024; 79(1): 39–48. doi: 10.1097/HEP.0000000000000529.
44. Giannini EG, Pasta A, Calabrese F et al. Second-line treatment for patients with primary biliary cholangitis: a systematic review with network meta-analysis. Liver Int 2025; 45(1): e16222. doi: 10.1111/liv.16222.
45. Colapietro F, Gershwin ME, Lleo A. PPAR agonists for the treatment of primary biliary cholangitis: old and new tales. J Transl Autoimmun 2023; 6: 100188. doi: 10.1016/ j.jtauto.2023.100188.
46. SPC Iqirvo 2025. Souhrn údajů o přípravku. 2025 [online]. Dostupné z: https: //www.ema.europa.eu/cs/documents/product-information/iqirvo-epar-product-information_cs.pdf.
47. Kowdley KV, Bowlus CL, Levy C et al. Efficacy and safety of elafibranor in primary biliary cholangitis. N Engl J Med 2024; 390(9): 795–805. doi: 10.1056/NEJMoa2306185.
48. Bachs L, Pares A, Elena M et al. Effects of long-term rifampicin administration in primary biliary cirrhosis. Gastroenterology 1992; 102(6): 2077–2080. doi: 10.1016/0016-5085(92)90335-v.
49. European Association for the Study of the Liver. EASL Clinical Practice Guidelines on liver transplantation. J Hepatol 2024; 81(6): 1040–1086. doi: 10.1016/j.jhep.2024.07.032.
50. Singal AK, Guturu P, Hmoud B et al. Evolving frequency and outcomes of liver transplantation based on etiology of liver disease. Transplantation 2013; 95(5): 755–760. doi: 10.1097/TP.0b013e31827afb3a.
51. Milkiewicz P. Liver transplantation in primary biliary cirrhosis. Clin Liver Dis 2008; 12(2): 461–472. doi: 10.1016/j.cld.2008.02.015.
52. Montano-Loza AJ, Hansen BE, Corpechot C et al. Factors associated with recurrence of primary biliary cholangitis after liver transplantation and effects on graft and patient survival. Gastroenterology 2019; 156(1): 96.e1–107.e1. doi: 10.1053/j.gastro.2018.10.001.